
Nhóm tác giả: ThS. Hoàng Thanh Trung – BS. Trương Đức Anh – TS.BS. Nguyễn Đỗ Ngọc Linh – TS.BS. Trần Ngọc Tuấn
Thiếu hụt 5-Alpha-Reductase là gì?
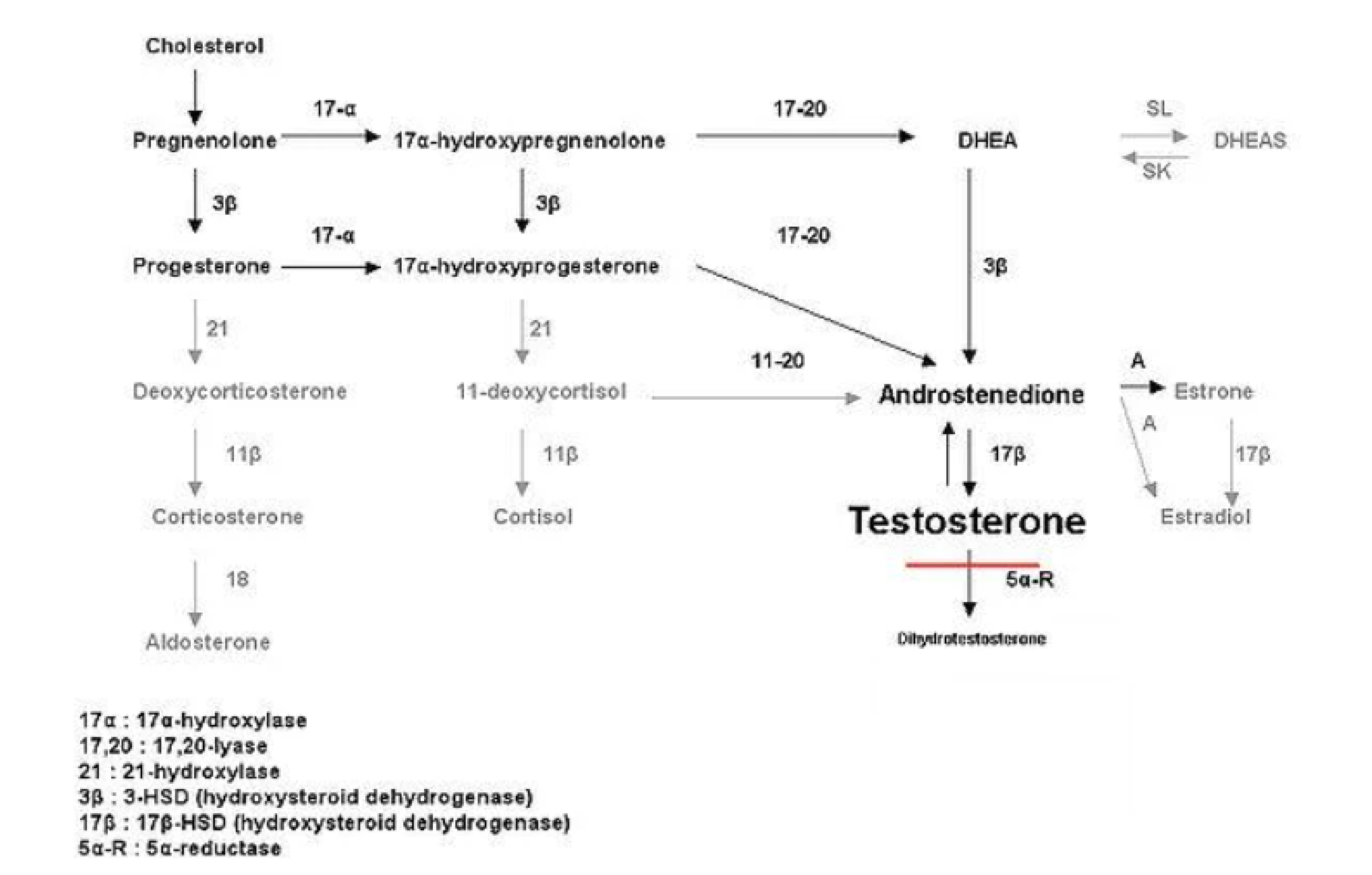
Sự thiếu hụt 5-Alpha-reductase loại 2 (5-ARD) là một rối loạn lặn trên nhiễm sắc thể thường, dẫn đến việc không thể chuyển đổi testosterone thành dihydrotestosterone (DHT) – là hormone có hoạt tính sinh lý cao hơn, đóng vai trò quan trọng trong quá trình nam tính hóa bình thường của cơ quan sinh dục ngoài ngay khi trẻ còn trong tử cung. Vì vậy, những bé trai với 46,XY bị thiếu hụt 5-alpha-reductase loại 2 được sinh ra với cơ quan sinh dục không rõ ràng, tức rối loạn phát triển cơ quan sinh dục với 46,XY (Disorder of Sexual Development = DSD).
Sơ đồ dưới đây thể hiện ảnh hưởng sinh hóa của sự thiếu hụt 5-Alpha-Reductase loại 2 trong quá trình sinh tổng hợp testosterone: mức độ testosterone tăng cao, trong khi mức độ dihydrotestosterone giảm đáng kể, dẫn đến tình trạng nam tính hóa dưới mức bình thường.
Những biểu hiện của thiếu hụt 5-Alpha Reductase
Bệnh nhân thiếu hụt 5-Alpha-Reductase loại 2 có biểu hiện kinh điển là cơ quan sinh dục không rõ ràng, dương vật giống như âm vật, bìu có hai chân, giả lỗ tiểu quanh âm đạo và tuyến tiền liệt sơ khai. Trong trường hợp khác, bệnh nhân trông nam tính hơn; có thể không có lỗ âm đạo riêng biệt, có lỗ tiểu riêng biệt ở dương vật hoặc thậm chí có niệu đạo dương vật.
Do sự bài tiết bình thường của yếu tố ức chế müllerian, bệnh nhân sẽ không có tử cung và ống dẫn trứng. Tinh hoàn còn nguyên vẹn và thường nằm trong ống bẹn hoặc bìu; tuy nhiên, đôi khi tinh hoàn nằm trong bụng (tinh hoàn ẩn). Sự biệt hóa của ống Wolffian là bình thường với túi tinh, biệt hóa ống dẫn tinh, mào tinh hoàn và ống phóng tinh. Tuyến tiền liệt nhỏ, không sờ thấy được và thô sơ ở tuổi trưởng thành.
Tăng sản tuyến tiền liệt lành tính (Benign Prostate Hyperplasia = BPH) và ung thư tuyến tiền liệt (Prostate Cancer = PC) đều không được báo cáo ở những bệnh nhân này.
Nguyên nhân của sự thiếu hụt 5-Alpha-Reductase loại 2?
Nguyên nhân của sự thiếu hụt 5-Alpha-Reductase loại 2 là sự thiếu hụt isozyme loại 2 của 5-Alpha-Reductase. Giống như hầu hết các rối loạn enzyme đơn lẻ, thiếu hụt 5-Alpha-Reductase loại 2 là một đặc điểm lặn trên nhiễm sắc thể thường và giới tính bị giới hạn bởi hội chứng lâm sàng chỉ ảnh hưởng đến nam giới di truyền, với các biểu hiện kiểu hình rất tinh tế ở nữ giới đồng hợp tử.
Ba gen mã hóa cho 5-Alpha-Reductase đã được xác định, mỗi gen mã hóa một loại isoenzyme hơi khác nhau. Chỉ các gen mã hóa cho Steroid 5-Alpha-Reductase loại 1 (SRD5A1) và Steroid 5-Alpha-Reductase loại 2 (SRD5A2) mới thích hợp để chuyển đổi testosterone thành DHT, trong khi gen mã hóa 5-Alpha-Reductase loại 3 không liên quan đến bất kỳ rối loạn phát triển giới tính nam nào (có thể liên quan đến rối loạn glycosyl hóa hiếm gặp).
Cả hai gen thích hợp, SRD5A1 và SRD5A2, chứa năm exon được phân tách bằng bốn intron. Phần lớn các đột biến đã được quan sát thấy ở exon 1 và 4.
Gen của SRD5A1 nằm trên dải 5p15. Sản phẩm của nó chỉ được biểu hiện ở da và gan ngoài vùng sinh dục ở mức độ thấp từ lúc 3 tuổi cho đến tuổi dậy thì, tại thời điểm này thì biểu hiện của enzyme có thể đo được ở tuyến bã nhờn và da đầu.
Gen của SRD5A2 đã được xác định nằm trên dải 2p23. Biểu hiện của gen này tương quan về mặt lâm sàng với các triệu chứng thiếu hụt 5-Alpha-Reductase loại 2. Ở tuổi trưởng thành, isoenzyme 5-Alpha-Reductase loại 2 được biểu hiện ở mức độ cao tại tuyến tiền liệt, da bộ phận sinh dục, mào tinh hoàn (epididymis), túi tinh (seminal vesicle) và gan.
Phân tích mối liên kết (Linkage Analysis) đã chứng minh rằng enzyme loại 1 không liên quan đến hội chứng lâm sàng của sự thiếu hụt 5-Alpha-Reductase loại 2. Điều đáng chú ý là quá trình nam hóa một phần ở nam giới bị thiếu hụt SRD5A2 xảy ra ở tuổi dậy thì, có thể do sự gia tăng hoạt động hoặc biểu hiện của enzyme loại 1 tại thời điểm đó.
Có 129 biến thể alen khác nhau trong gen SRD5A2 ở những người bị thiếu hụt 5-Alpha-Reductase. Các biến thể là missense mutations (n = 83); small deletions (n = 12); splicing mutations (n = 6); stop codons (n = 4); small indels (n = 20) và gross deletions (n = 4).
Chẩn đoán phân biệt (Differential diagnoses)
• 17-Beta-Hydroxysteroid Dehydrogenase Deficiency (Thiếu hụt 17-Beta-Hydroxysteroid Dehydrogenase)
• 17-Hydroxylase Deficiency Syndrome (Hội chứng thiếu hụt 17-Hydroxylase)
• 3-Beta-Hydroxysteroid Dehydrogenase Deficiency (Thiếu hụt 3-Beta-Hydroxysteroid Dehydrogenase)
• Adrenal Hypoplasia (Tăng sản tuyến thượng thận)
• Androgen Insensitivity Syndrome (Hội chứng tinh hoàn nữ tính hóa)
• Congenital Adrenal Hyperplasia (Tăng sản tuyến thượng thận bẩm sinh)
• Denys-Drash Syndrome (Hội chứng Denys-Drash)
• Differences (Disorders) of Sex Development (Rối loạn phát triển giới tính)
• Gonadal Dysgenesis
• Hypopituitarism (Panhypopituitarism)
• Kallmann Syndrome and Idiopathic Hypogonadotropic Hypogonadism (Hội chứng Kallman và Hội chứng suy sinh dục vô căn)
• LH Receptor Defect
• Microphallus (Hội chứng dương vật nhỏ)
• Ovotesticular Disorder of Sexual Development
• P450 Oxidoreductase Deficiency (Thiếu hụt P450 Oxidoreductase)
• Smith-Lemli-Opitz Syndrome (Hội chứng Smith-Lemli-Opitz)
• STAR Deficiency
Chẩn đoán
Tại FAMILY, chúng tôi sẽ thực hiện:
• Karyotype/Array/SHPT with specific X and Y probes.
(Karyotype/Array/SHPT với đầu dò X và Y riêng biệt)
• 17-hydroxyprogesterone, follicle-stimulating hormone (FSH), luteinizing hormone (LH), testosterone, dihydrotestosterone (DHT), anti-Müllerian hormone (AMH), electrolytes, and urinalysis.
• Abdominopelvic ultrasound to assess for the presence of internal male/female structures.
(Siêu âm bụng để đánh giá sự hiện diện của các cấu trúc nam/nữ bên trong)
Điều trị
Chúng tôi sẽ không nói chi tiết tại đây về việc điều trị chứng rối loạn này, bao gồm cả liệu pháp hormone (2% dihydrotestosterone (DHT) cream, testosterone enanthate or cypionate (Delatestryl), estrogens, conjugated (Premarin), ethinyl estradiol (Estinyl)).
SOURCES
[Guideline] Houk CP, Lee PA. Consensus statement on terminology and management: disorders of sex development. Sex Dev. 2008. 2(4-5):172-80.
Silver RI, Russell DW. 5alpha-reductase type 2 mutations are present in some boys with isolated hypospadias. J Urol. 1999 Sep. 162(3 Pt 2):1142-5.
Imperato-McGinley J, Zhu YS. Androgens and male physiology the syndrome of 5alpha-reductase-2 deficiency. Mol Cell Endocrinol. 2002 Dec 30. 198(1-2):51-9.
Zhu YS, Imperato-McGinley JL. 5 alpha-reductase isozymes and androgen actions in the prostate. Ann N Y Acad Sci. 2009 Feb. 1155:43-56.
Choi JH, Kim GH, Seo EJ, Kim KS, Kim SH, Yoo HW. Molecular analysis of the AR and SRD5A2 genes in patients with 46,XY disorders of sex development. J Pediatr Endocrinol Metab. 2008 Jun. 21(6):545-53.
Okeigwe, I. and Kuohung, W. 5-Alpha Reductase Deficiency: A 40-year Retrospective Review. Curr Opin Endocrinol Diabetes Obes. 2014. 21:483-487.
Russell DW, Wilson JD. Steroid 5 alpha-reductase: two genes/two enzymes. Annu Rev Biochem. 1994. 63:25-61.
Thigpen AE, Davis DL, Milatovich A, Mendonca BB, Imperato-McGinley J, Griffin JE. Molecular genetics of steroid 5 alpha-reductase 2 deficiency. J Clin Invest. 1992 Sep. 90(3):799-809.
Azzouni F, Godoy A, Li Y, Mohler J. The 5 alpha-reductase isozyme family: a review of basic biology and their role in human diseases. Adv Urol. 2012. 2012:530121.
Maimoun L, Philibert P, Bouchard P, Ocal G, Leheup B, Fenichel P. Primary amenorrhea in four adolescents revealed 5a-reductase deficiency confirmed by molecular analysis. Fertil Steril. 2011 Feb. 95(2):804.e1-5.
Costa EM, Domenice S, Sircili MH, Inacio M, Mendonca BB. DSD due to 5a-reductase 2 deficiency – from diagnosis to long term outcome. Semin Reprod Med. 2012 Oct. 30(5):427-31.
Batista RL, Mendonca BB. Integrative and analytical review of the 5-alpha-reductase type 2 deficiency worldwide. Appl Clin Genet. 2020. 13:83-96. [QxMD MEDLINE Link]. [Full Text].
Medonca BB, Domenice S, Arnhold, JP, and Costa EMF. 46, XY disorders of sex development (DSD). Clinical Endocrinology. 2009. 70:173-187.
Allen L. Disorders of Sexual Development. Obstet Gynecol Clin N Am. 2009. 36:25-45.








